Bite-sized learning:Understanding Familial Hypercholesterolemia
Michaela Nuttall RGN MSc, Founder & Director, Learn With NursesJoanne Haws RN MSc, Clinical Director...
Michaela Nuttall RGN MSc, Founder & Director, Learn With NursesJoanne Haws RN MSc, Clinical Director, Learn With Nurses
Practice Nurse 2024;54(2):10-11
Familial hypercholesterolemia (FH) is a genetic disease which results in the reduced clearance of atherogenic LDL-cholesterol, often referred to as bad cholesterol, and therefore people are at significantly increased risk of premature heart disease.
WHAT’S THE PROBLEM?
FH is one of the most common inherited conditions, and in the UK, the prevalence is estimated to be 1 in 250 people. So, about 220,000 people in the UK have FH, yet less than 8% are currently identified.1
People with FH have elevated LDL cholesterol levels from birth, and the process of atherosclerosis, blockages in arteries development, starts early in life. As a result of their FH, the incidence of fatal or non-fatal myocardial infarction without treatment is about 50% by the age of 50 years in men and about 30% by the age of 60 years in women.2
A typical GP practice with 10,000 patients might have up to 40 patients with FH and many of these are likely to have not been identified or, if identified, are not being offered high-intensity statin therapy or other lipid management medication.
GENES INVOLVED WITH FH3
1. LDL receptor genes: This is the most common gene fault. There are not enough LDL receptors, meaning cholesterol builds up in the blood.
2. APOB gene (apolipoprotein B): Two or three out of every 100 people with FH have a fault with this gene. It means LDL cholesterol can’t bind well to LDL receptors. So LDL is taken out of the blood too slowly, and cholesterol remains elevated.
3. PCSK9 gene: Only a few people have this type of FH. This fault means that LDL receptors are broken down in the liver, so they can’t remove cholesterol from the blood.
Heterozygous FH – gene from one parent
Homozygous FH – gene from both parents (very rare)
DETECTION
There are a variety of ways FH can be detected.4
Opportunistic
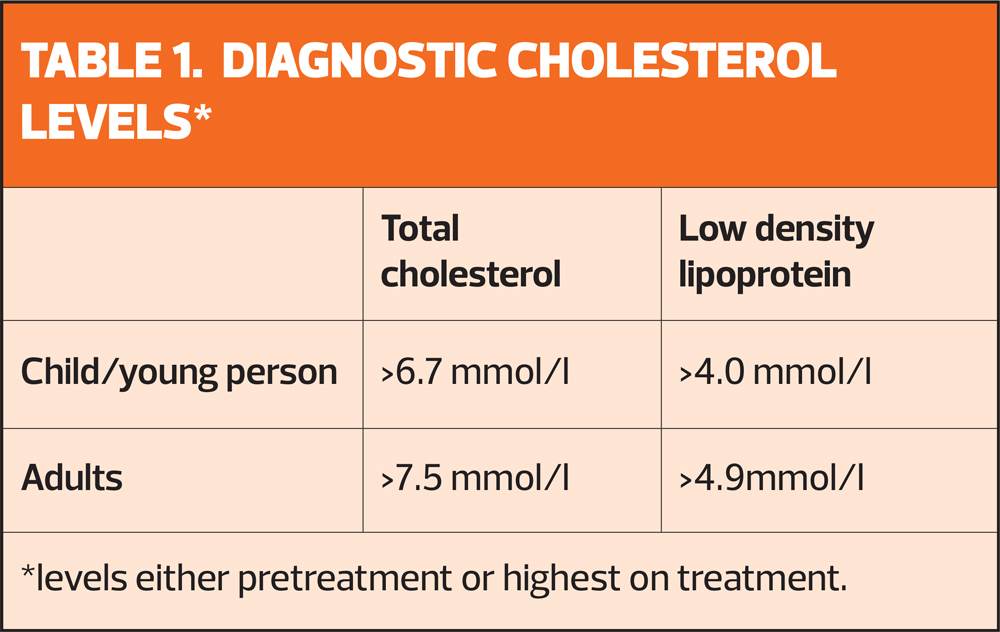
Suspect FH in adults with a total cholesterol level greater than 7.5 mmol/l or a personal or family history of premature coronary heart disease (an event before 60 years in first-degree relative)
For people with a personal or family history of premature coronary heart disease but whose total cholesterol is unknown, offer to measure their total cholesterol.
Systematic
Search primary care records for people younger than 30 years, with a total cholesterol concentration greater than 7.5 mmol/l and 30 years or older, with a total cholesterol concentration greater than 9.0 mmol/l as these are the people who are at highest risk of FH.
Cascade testing
Relatives of someone with FH should have a DNA test. Refer to an FH specialist. They may also request cholesterol testing in primary care.
DIAGNOSIS
Before a diagnosis of FH is considered, exclude secondary causes of hypercholesterolaemia, including uncontrolled diabetes mellitus, obesity, excess alcohol consumption, untreated hypothyroidism and some medications, for example, thiazide diuretics and ciclosporin.
Then, the patient should be referred to a lipid clinic where the Simon Broome criteria are often used for diagnosis:
Simon Broome Criteria
Definite familial hypercholesterolaemia (FH) if:
- cholesterol concentrations as defined in table 1 and tendon xanthomas, or
evidence of these signs in first- or second-degree relative, or
- DNA-based evidence of an LDL-receptor mutation, familial defective apo B100, or a PCSK9 mutation.
Possible FH if they have cholesterol concentrations as defined in table 1 and at least one of the following:
- Family history of myocardial infarction: aged younger than 50 years in second-degree relative or aged younger than 60 years in first-degree relative.
- Family history of raised total cholesterol, greater than 7.5 mmol/l in adult first- or second-degree relative or greater than 6.7 mmol/l in child, brother or sister aged younger than 16 years.
MANAGEMENT
All patients with confirmed FH should be offered a high-intensity statin, or if contraindicated, ezetimibe. Initial management usually occurs in lipid clinics, and depending on the severity of FH, care might be passed back to primary care. Homozygous FH should always be managed by a specialist centre.
All people with FH should be offered individualised nutritional advice from a healthcare professional with specific expertise in nutrition and it is recommended:
- Total fat intake is 30% or less of total energy intake
- Saturated fats are 10% or less of total energy intake
- Intake of dietary cholesterol is less than 300 mg/day
- Saturated fats are replaced by increasing the intake of monounsaturated and polyunsaturated fats.
REVIEW (IF BEING FOLLOWED UP IN PRIMARY CARE)
- Review lipid levels
- Medication review including adherence
- Weight/BMI
- Review of CVD risk factors and any support required
- Review of overall health and wellbeing, including mental health
- Consider support for relatives who may be undergoing cascade or cholesterol testing
See also, Reducing cardiovascular disease: latest recommendations
References
1. NHS England. Familial Hypercholesterolemia (FH) https://www.england.nhs.uk/london/london-clinical-networks/our-networks/cardiac/familial-hypercholesterolaemia/
2. Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168(1):1–14.
3. Heart UK. What is familial hypercholesteraemia. https://www.heartuk.org.uk/fh/what-is-fh
4. NICE CG71. Familial hypercholesterolaemia: identification and management https://www.nice.org.uk/guidance/cg71

Related articles
View all Articles