Haematology for the practice nurse
Dr Mary Lowth
Dr Mary Lowth
MA, MB, BChir, FRCGP
GP and medical educator
An enormous number of acute and chronic conditions can affect the structure and function of the cellular and protein components of the blood. It is impossible to cover all of haematology in one article, but this gives an overview of primary conditions of the blood, focusing particularly on more serious haematological disease
Many haematological conditions are relatively common, but can present so insidiously, and with such non-specific symptoms that patients themselves may not notice anything is wrong. The practice nurse therefore needs to be both alert to unexplained abnormalities on blood tests, even mild ones on routine tests, and aware of the possible significance of vague symptoms, particularly those that persist.
COMPONENTS OF THE BLOOD
The main cell types in blood are:
- Red blood cells
- White blood cells
- Platelets.
Most are produced in the bone marrow, although T and B lymphocytes are also produced in the lymph nodes and spleen, and T cells in the thymus gland. All blood cells originate from undifferentiated stem cells, which divide first into immature red or white blood cells, or platelets, then divide further to become the final, functional cell.
White blood cells, whose main role is to fight infection and ingest abnormal cells, then last for a few hours to a few days. Platelets, whose role is in the clotting mechanism, last about 10 days and red cells, whose main role is to carry oxygen to tissues, about 120 days. The bone marrow produces more white blood cells in response to infections and injuries, more red cells in response to blood loss or lowered oxygen levels, and more platelets in response to bleeding.
The haematological disorders discussed here are mainly those that affect these cells and their production and function.
DISORDERS OF HAEMOGLOBIN AND RED CELLS
These include abnormalities of shape, production and destruction of red cells, and abnormalities of the haemoglobin they carry.
Anaemia
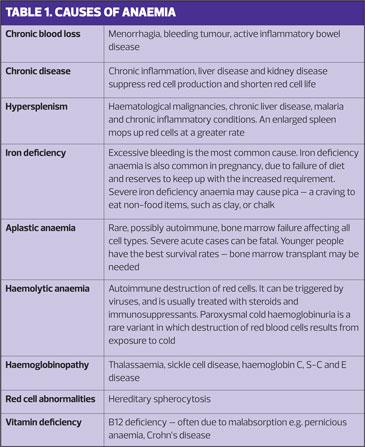
This is the most common red cell disorder encountered in primary care. Crucially, this is not a diagnosis but a red flag symptom that requires one.
Anaemia may result from blood loss or inadequate production of red cells or haemoglobin. Unexplained anaemia is a red flag for serious disease, particularly in older patients where it may be the presenting symptom of cancer, particularly of the GI tract. This may be detected incidentally as a part of a long-term medication review.
Mild anaemia is often asymptomatic, although more severe anaemia causes tiredness, breathlessness and pallor, and sometimes jaundice. While rapid blood loss causes acute symptoms, it is possible to become extremely anaemic with relatively mild symptoms if the decline is slow and steady. Haemoglobin levels as low as 4mg/dl have been reported in patients still managing their normal daily lives.
Anaemia in women of menstrual age is commonly caused by menorrhagia, but it is essential that even this common diagnosis is confirmed by supportive history, examination and response to treatment.
Anaemia may also be haematological in origin, resulting from primary bone marrow disorders causing impaired production, including aplastic anaemia, myeloproliferative disorders and myelofibrosis. Causes of anaemia are listed in Table 1.
Polycythaemia
Polycythaemia is an excess of red cells. It results from increased red cell production, which may be caused by low oxygen levels (such as in chronic lung disease or after time at altitude) or by abuse of erythropoetin. Polycythaemia increases haematocrit, and the thicker blood may sludge in small vessels, risking stroke.
Polycythaemia vera (PV) is a rare myeloproliferative disorder, mainly affecting people aged over 40 years. It results from a bone marrow malignancy affecting red cell precursors, resulting in the overproduction of red cells, which may eventually crowd out other cell production.
Haemoglobinopathies
These are lifelong genetic disorders affecting the quantity or shape of the haemoglobin molecule. Most common are thalassaemia, in which haemoglobin is deficient, and sickle cell disease and trait, in which it is misshapen.
Thalassaemia
Thalassaemia is most common in those of Mediterranean, South Asian, Southeast Asian and Middle Eastern origin. Severe disease can be inherited if both parents carry the trait, so antenatal screening is offered to all women in the UK.
Thalassaemia major causes marked anaemia requiring frequent transfusions and leading to bone problems, splenic enlargement, and iron overload causing heart and liver disease. Splenectomy may be needed.
Minor thalassaemia (thalassaemia trait) is usually asymptomatic. It causes a mild microcytic anaemia that resembles iron deficiency but does not respond to iron. Its prime importance for the practice nurse lies in the need to suspect it as a possible cause of anaemia, and to facilitate pre-conceptual counselling and testing of those affected.
Sickle cell disease
In sickle cell disease abnormal haemoglobin affects the shape of red cells, which causes them to collapse in low oxygen levels, usually in small blood vessels. It mainly affects people of African, Caribbean, Middle Eastern, Eastern Mediterranean and Asian ethnicity. The full disease is inherited from both patients, and causes anaemia and very painful sickling episodes.
Patients with sickle cell trait have only one faulty gene and are usually asymptomatic, with mild anaemia that does not respond to iron therapy.
Red cell abnormalities
Several uncommon inherited conditions affect red cell shape; the best known is hereditary spherocytosis (HS), in which the red cells are spherical rather than disc-shaped, and red cell life span is significantly reduced. HS typically causes splenomegaly, which may necessitate splenectomy, and can cause anaemia and jaundice.
DISORDERS OF PLATELETS AND CLOTTING MECHANISMS
The clotting mechanism of the blood has several elements and abnormalities of any may leading to problems with bleeding or clotting. These include fragile blood vessels (caused by some diseases such as amyloidosis), altered platelet number or function, and problems with the cascade of proteins (clotting factors) that lead to clot formation.
Clotting factor disorders
Clotting factors are made in the liver. Hereditary deficiencies include:
- Haemophilia – factor VIII deficiency
- Christmas Disease – factor IX deficiency
- von Willebrand’s disease – deficiency of a protein which helps activate Factor VIII.
Impaired clotting can also result from severe liver disease, vitamin K deficiency and from an autoimmune attack on clotting factors, preventing their function.
Clotting factor deficiencies can lead to heavy or unusual bleeding, typically after tissue injury or surgery, and bleeding into joints. Treatment is usually by replacement of the factors involved, although this has in the past led to the iatrogenic infection of patients with HIV and hepatitis C through use of pooled, contaminated blood products.
Thrombophilia is an increased tendency to form blood clots. Mutations of some clotting factors increase clotting tendencies. The Factor V Leiden mutation causes an increased tendency to thrombosis during pregnancy or when taking oestrogen. Inherited deficiencies of the natural anticoagulants protein S, protein C and antithrombin also increase the risk of thrombosis. The risk is then compounded in hypercoagulable states such as pregnancy and after surgery.
PLATELET DISORDERS
Low platelet levels
Thrombocytopenia (low platelet count) results from decreased platelet production or increased destruction. Very low platelet levels typically lead to increased or prolonged bleeding from mucous membranes such as the nose and gums. More severe deficiency can produce deeper, life-threatening bleeds. Normal platelet levels are 150,000-450,000 per dl. There is a lot of spare capacity, but levels less than 10,000 put patients at severe risk.
Production failure may occur in myeloproliferative disorders and haematological malignancies, in myelodysplasia and in aplastic anaemia, in which functional marrow is lost or replaced by abnormal tissue. It may also occur as a medication side effect, or after exposure to some chemicals.
Increased platelet breakdown is often autoimmune, and can occur in the presence of other autoimmune diseases or alone, when it is called idiopathic thrombocytopaenia purpura (ITP). ITP is the most common cause of platelet deficiency in children.
Some bacterial infections, including meningococcus, can cause destruction of platelets. Drugs, including heparin, quinine, and anticonvulsants, can cause platelet breakdown.
Raised platelet levels
Thrombocythaemia (raised platelet levels) can be a transient response to infection. Essential thrombocythaemia (ET) is a myeloproliferative disorder similar to polycythaemia vera, but in which the platelet cell line proliferates. The excess platelets may cause thrombosis, and can also collect in the spleen, leading to splenomegaly. ET can be a precursor to acute myeloid leukaemia, and a cause of myelofibrosis.
Table 2 summarises the symptoms of thrombocytopaenia and thrombocythaemia.
DISORDERS OF WHITE CELLS AND BONE MARROW
White cells include neutrophils, lymphocytes (B and T types), monocytes, eosinophils and basophils. We normally produce around a billion a day. Leukopaenia
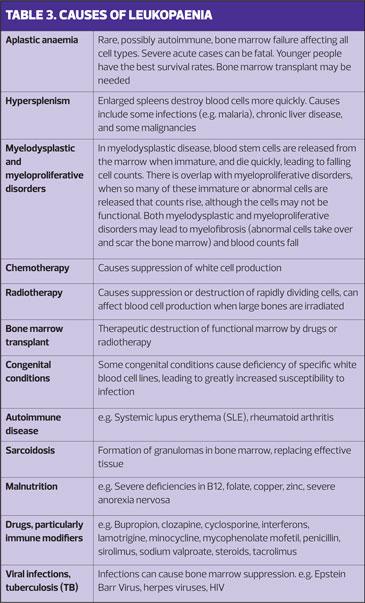
Deficiency of white cells, leukopenia, increases susceptibility to infection.
Examples of the many conditions that cause white cell production to fall are given in Table 3.
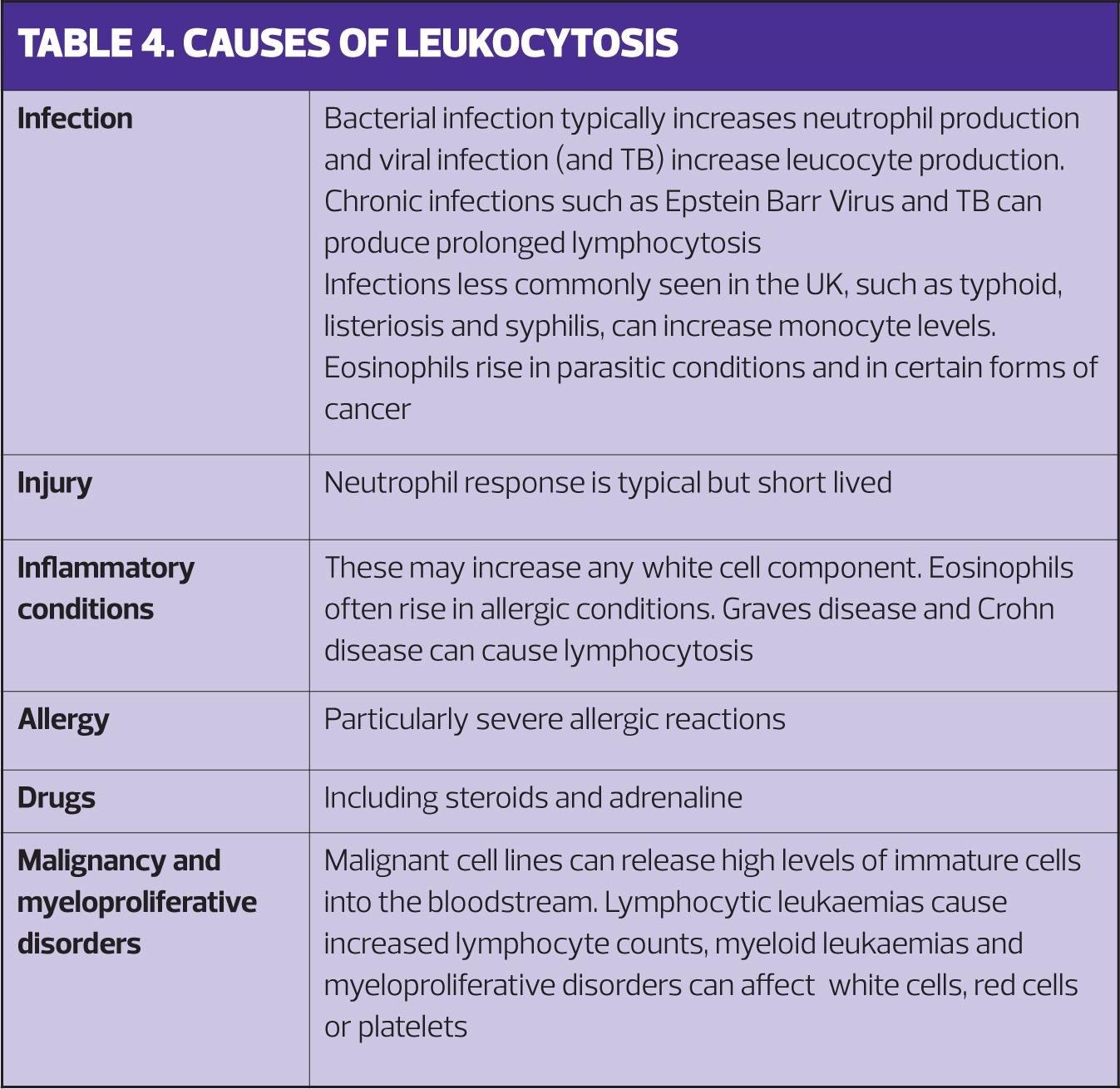
Leukocytosis
Increased production of white cells is a normal response to infection or injury, and can also be caused by some drugs, including steroids. Neutrophils and leucocytes are most commonly affected. However, increased white blood cell production is also caused by leukaemias; when white counts are extremely high acute neoplastic processes are a likely cause. Extremely high levels of neutrophils (> 100,000 per microlitre) can cause hyperviscosity severe enough to cause breathing difficulty and acute stroke. Causes of leucocytosis are given in Table 4.
HAEMATOLOGICAL MALIGNANCIES
The diagnosis of haematological malignancy represents perhaps the greatest challenge for the practice nurse, and the greatest opportunity for success. Diagnostic delay is common in haematological cancer.1 Prior to diagnosis, patients often have multiple primary care consultations without their condition being identified, although only about a third are asymptomatic at diagnosis.1
Diagnostic delay can increase complications and decrease treatment success, and there is an urgent need to improve diagnostic efficiency. This is not an easy area. Symptoms are non-specific and may creep on so quietly that patients don’t think to mention them, or may put them down to their age or fitness. Tiredness and pain are particularly common, although lymphadenopathy (in lymphoma) and bleeding and bruising (in acute leukaemia) may occur and are important, specific clues. Night sweats, weight loss and unexplained anaemia are also common and should always raise concerns – as should any persisting symptoms which do not resolve as expected.
Leukaemia
Leukaemias are cancers of the haematogenous tissues that produce immature, white blood cells, sometimes in high enough numbers to make the blood appear white (hence the name). Lymphocytic leukaemias affect lymphocytes (B cells or T cells) while myeloid leukaemias affects the myeloid line (which normally produces neutrophils, monocytes, eosinophils, red cells and platelets), producing immature cells called myeloblasts.
Leukaemias can be acute, with highly abnormal blood test results and many immature cells visible on peripheral blood films. They can also be chronic and insidious, with blood tests little altered and symptoms vague and non-specific. The symptoms of leukaemia are shown in Box 1. Previous radiotherapy and some types of chemotherapy increase the risk of all leukaemias.
There are four main types of leukaemia:
- Acute lymphocytic leukaemia (ALL) – the most common type of cancer in children, and prognosis with treatment is good. Adults tend to do less well with treatment. ALL is more common in those who have had a sibling with ALL
- Acute myeloid leukaemia (AML) – more common in people over 65 years of age, but prognosis is, again, better in children. People with some genetic disorders, such as Down syndrome, have an increased risk of AML and ALL
- Chronic lymphocytic leukaemia (CLL) – relatively common in older adults and may be present as a chronic disease for many years. CLL is more common in patients of white ethnicity
- Chronic myelogenous leukaemia (CML) – uncommon and typically affects older adults. Most cases result from chromosome changes in marrow precursors, resulting in an abnormal chromosome 22 (the Philadelphia chromosome). Men are more likely to develop AML and CML. Exposure to certain chemicals, such as benzene, increases the risk of myeloid leukaemias.
Treatment depends on which cell is proliferating, and on whether the leukaemia is acute or chronic. Treatments may include chemotherapy, targeted therapies such as monoclonal antibodies, Chimeric Antigen Receptors Cell Therapy (CAR-T), other drug therapies and bone marrow transplant.
Treatment of acute leukaemias is approached in phases, which may include:
- Induction (to eliminate most abnormal cells)
- Consolidation (to destroy remaining abnormal cells)
- Maintenance (to prevent recurrence), and
- Prevention (in ALL preventative treatment is given to the central nervous system to prevent malignant cells from taking refuge there.)
Treatment of chronic leukaemias depends on the level and nature of abnormal cells. Early stage CLL usually requires only monitoring, although progressive disease treatment may involve any of the therapies used for acute leukaemia. CML, however, benefits from early treatment at diagnosis.
LYMPHOMA
Lymphomas are cancers of the lymphatic system, which includes the lymph nodes, spleen, thymus and bone marrow. They are more common with greater age, particularly in men. Some infections, including HIV and Epstein Barr Virus, increase the risk of lymphoma, as does prolonged use of immunosuppressant drugs. Lymphomas are most often solid tumours that often present with a large painless lymph node, typically in the neck. They may not always cause blood tests to be deranged.
There are many types of lymphoma, but the most common types are Hodgkin’s lymphoma and Non-Hodgkins lymphoma. These differ in cell type; in Hodgkin’s lymphoma a specific type of abnormal cell called a Reed-Sternberg cell is present. Lymphoma is common in patients over 55, but Hodgkin’s Lymphoma also occurs relatively commonly in younger adults. Typical symptoms of lymphoma are shown in Box 2.
Waldenstrom’s macroglobulinemia is a rare subtype in which excessive, large, non-functional immunoglobulins are produced by the abnormal cells. Symptoms resemble those of other lymphomas, but with additional effects of hyperviscosity, which can cause peripheral numbness and tingling, headache, altered vision, confusion and stroke.
Treatment of lymphoma depends on the stage and type of the disease, and may involve chemotherapy, radiotherapy, other drug agents and bone marrow transplant. New targeted therapies are currently undergoing clinical trials.
Myelofibrosis
Myelofibrosis is a condition that can result from leukaemias or myeloproliferative disorders, in which the bone marrow is first disrupted then eventually replaced by scar tissue. It can be asymptomatic for years. Eventual symptoms resemble those of chronic leukaemias.
Myeloma and MGUS
Multiple myeloma is a cancer of plasma cells, which are fully matured B-lymphocytes. These proliferate and produce abnormal monoclonal proteins, often of one single type, which can damage other organs, particularly the kidneys.
Myeloma is an uncommon cancer seen almost exclusively in patients over 60 years old. It often has a slow course, over years, but once symptomatic, tends to cause bone and kidney symptoms, including bone pain, pathological fractures and kidney stones. The abnormal proteins can cause acute renal impairment. Calcium levels are often raised and cause nausea, constipation, thirst and confusion. Other symptoms resemble those of leukaemias, with fatigue very common, and frequent infections resulting from the replacement of healthy bone marrow components.
Myeloma is usually preceded by an asymptomatic and relatively common condition called Monoclonal Gammopathy of Undetermined Significance (MGUS). About 1 in 100 people with MGUS will develop myeloma each year.
Myeloma is more common in men, in patients of black ethnicity and in those with a close family history of the disease. It is one of the important causes of an unexplained, very raised ESR.
Myeloma is diagnosed on bone marrow testing, and may be treated with targeted therapies, biological or chemotherapy agents, steroids, radiotherapy and, sometimes, bone marrow transplant.
STAYING ALERT FOR HAEMATOLOGICAL MALIGNANCY
Red flag symptoms
Haematological malignancies become more common with increasing age, but are also among the more common childhood cancers. While many cause non-specific symptoms that can creep on so slowly that patients don’t think them significant, it is the role of health professionals to try to explain all symptoms. If our explanations don’t work, because symptoms do not resolve as expected, we need to investigate further.
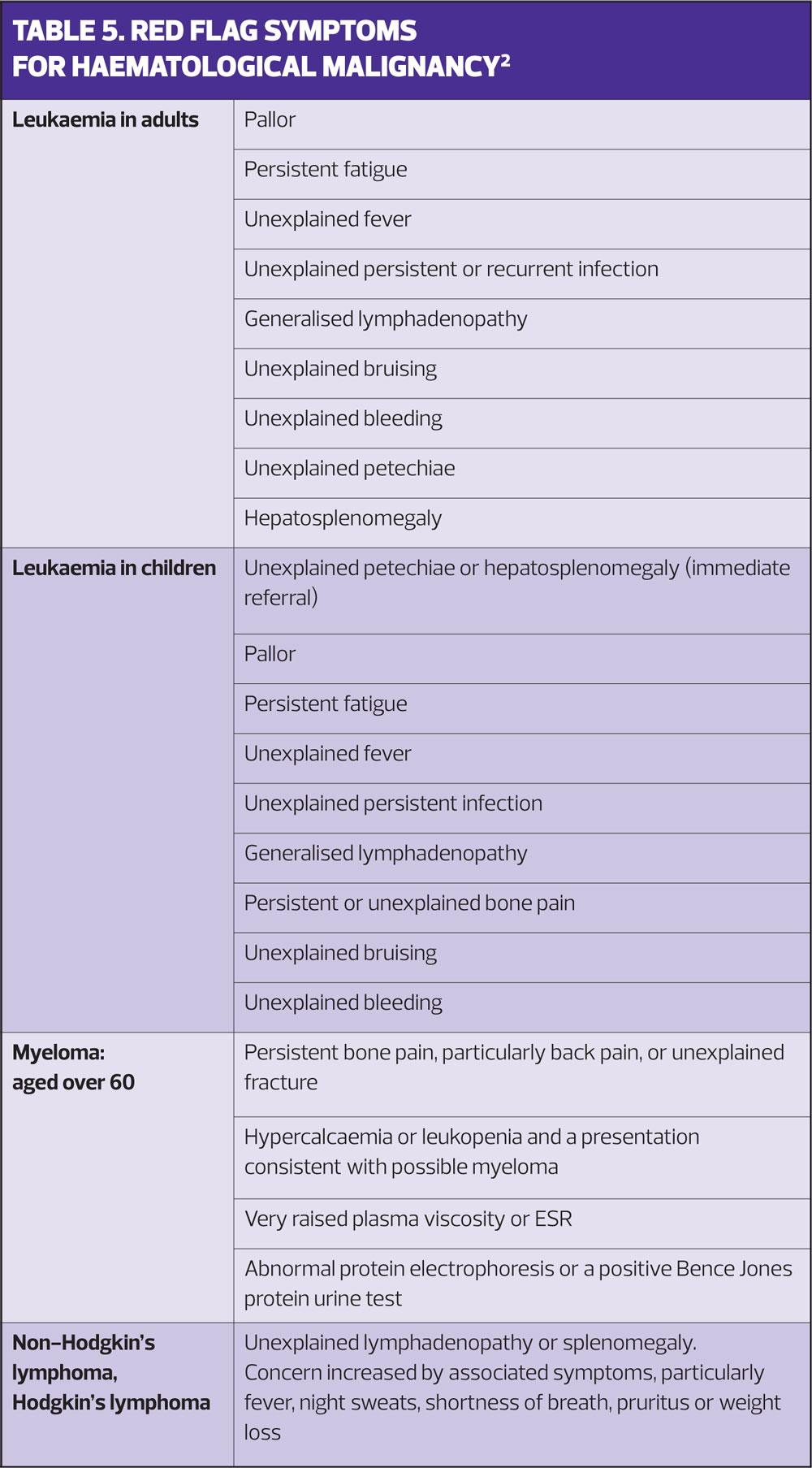
Table 5 lists the NICE red flag and other symptoms of haematological malignancy.2 These generally merit urgent, sometimes immediate investigation. In children this may mean urgent admission.
RESEARCH AND NEW DEVELOPMENTS
Haematological malignancy is a busy area for pharmacological research. There are ongoing trials of treatments for leukaemias, particularly CLL, and in monoclonal antibody therapies for myeloma.
One treatment that received much attention recently is CAR-T, (marketed as Kymriah),3 which modifies a patient’s own immune cells; T-cells are primed by the therapy to recognise and destroy cancer cells. CAR-T therapy is risky but can be curative where other treatments have failed. It is a one-time, hospital-administered treatment. In September NICE approved it for B-cell acute lymphoblastic leukaemia (ALL) in young people up to age 25 years, in one of the fastest funding approvals in the history of the NHS.3 CAR-T has been described as a game-changer and is also used for some adult B cell lymphomas.
SUMMARY
Haematological disease is a wide subject area and symptoms are often vague or non-specific. The onset of symptoms can be insidious and patients may feel they are not worth mentioning. As health professionals our role is to find an explanation for those symptoms that people present with, but we also owe it to our patients to investigate further if those symptoms fail to resolve as we expect.
REFERENCES
1. Howell DA, Smith AG, Jack A, et al. Time-to-diagnosis and symptoms of myeloma, lymphomas and leukaemias: a report from the Haematological Malignancy Research Network. BMC Hematology. 2013;13(1):9. doi:10.1186/2052-1839-13-9
2. NICE Guideline NG12: Suspected Cancer: Recognition and Referral, June 2015. https://www.nice.org.uk/guidance/ng12/chapter/1-Recommendations-organised-by-site-of-cancer#haematological-cancers
3. NICE Technology Appraisal 554: Tisagenlecleucel for treating relapsed or refractory B-cell acute lymphoblastic leukaemia in people aged up to 25 years, Dec 2018. www.nice.org.uk